In the Allen lab we work to make our code freely available we actively support and update a few pipelines and software listed below. We principally aim to develop software to help answer questions in biodiversity and solve problems in biology.
Label_Reconciliations: This code will reconcile three different transcripts into on ‘best guess transcript’ taking into account mostly majority rules and fuzzy matching. It is most commonly used on public volunteer data of transcripts from museum specimens from Notes from Nature.
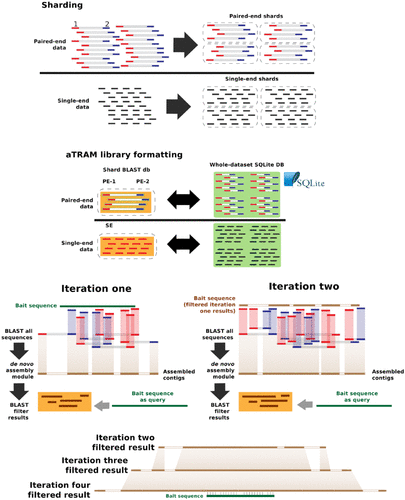
aTRAM: automated Target Restricted Assembly Method – all code and menus can be found here. aTRAM is an iterative assembler that performs reference-guided local de novo assemblies using a variety of available methods. It is well-suited to various tasks where Next-Generation Sequence (NGS) data needs to be queried for gene sequences, such as phylogenomics.
Other pipelines can be found on my Github.
